
The systemic impurity concept is an integrated analysis of any step in the production process assessing any compound/ reaction partner within the production process that could possibly be involved in the formation of an impurity or degradation product. This holistic analysis allows a reliable prediction of the structure of the unknown impurity, identification of the generation mechanism, and subsequently strategic impurity management.
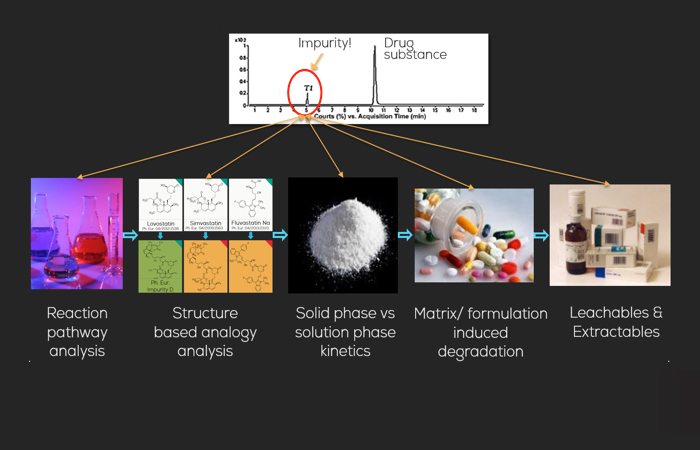
SYSTEM IMPURITY ANALYSIS
1. Analysis of each reaction step in the synthesis pathway of the drug substance
Raw material, reaction intermediates, by-products, reactants
2. Structure based analogy analysis
Evaluation of monographed drug substance impurities of certain members of a drug class allows analogy conclusions for other members of the same drug family.
3. Integral analysis of the entire production process
Excipient interactions, excipient impurities, matrix interactions, and pharmaceutical dosage form may result in incompatibility of drug substance and excipient composition.
4. Solid phase vs solution phase kinetics
Impurities may result from reactions in solution phase as well as in solid phase, often with higher reaction kinetics compared to the reaction in solution. Understanding of the mechanism behind the formation reaction allows in vitro generation of the impurity using heterogenous catalysis.
5. Packaging induced reaction products and stability issues
Leachables and Extractables (L&E) from filters, primary packaging material e.g. blisters, foil, tubes, etc. are impurities which may react with the drug substance forming potentially toxic compounds. Permeability properties (vapour, O2, etc.) strongly impact drug product stability.
6. Strategies for impurity optimization/ circumvention
Based on the assessment of the synthesis pathway and pharmaceutical development, recommendations for the prevention, reduction, and optimization are given.
7. Proposal of regulatory compliant argumentation lines
The impurity evaluation is integral part of the CTD (Common Technical Document) required for market authorisation of a drug product. Strategies and argumentation lines need to be developed to justify the presence of impurities and degradation products.